密度泛函理论基本
一些定义
波函数(wave function)
为复数函数,表示在空间位置和时间找到所描述粒子的概率幅,表示在相应时空位置找到该粒子的概率密度。因此,以一维空间中运动的粒子为例,粒子在 的概率为
P_{a \leq x \leq b}=\int_{a}^{b}|\Psi(x, t)|^{2} \mathrm{~d} x
显然,粒子在全空间上出现的概率为1(归一性),即
\int_{-\infty}^{\infty}|\Psi(x, t)|^{2} \mathrm{~d} x=1
下图是一维空间粒子波函数的两个示例
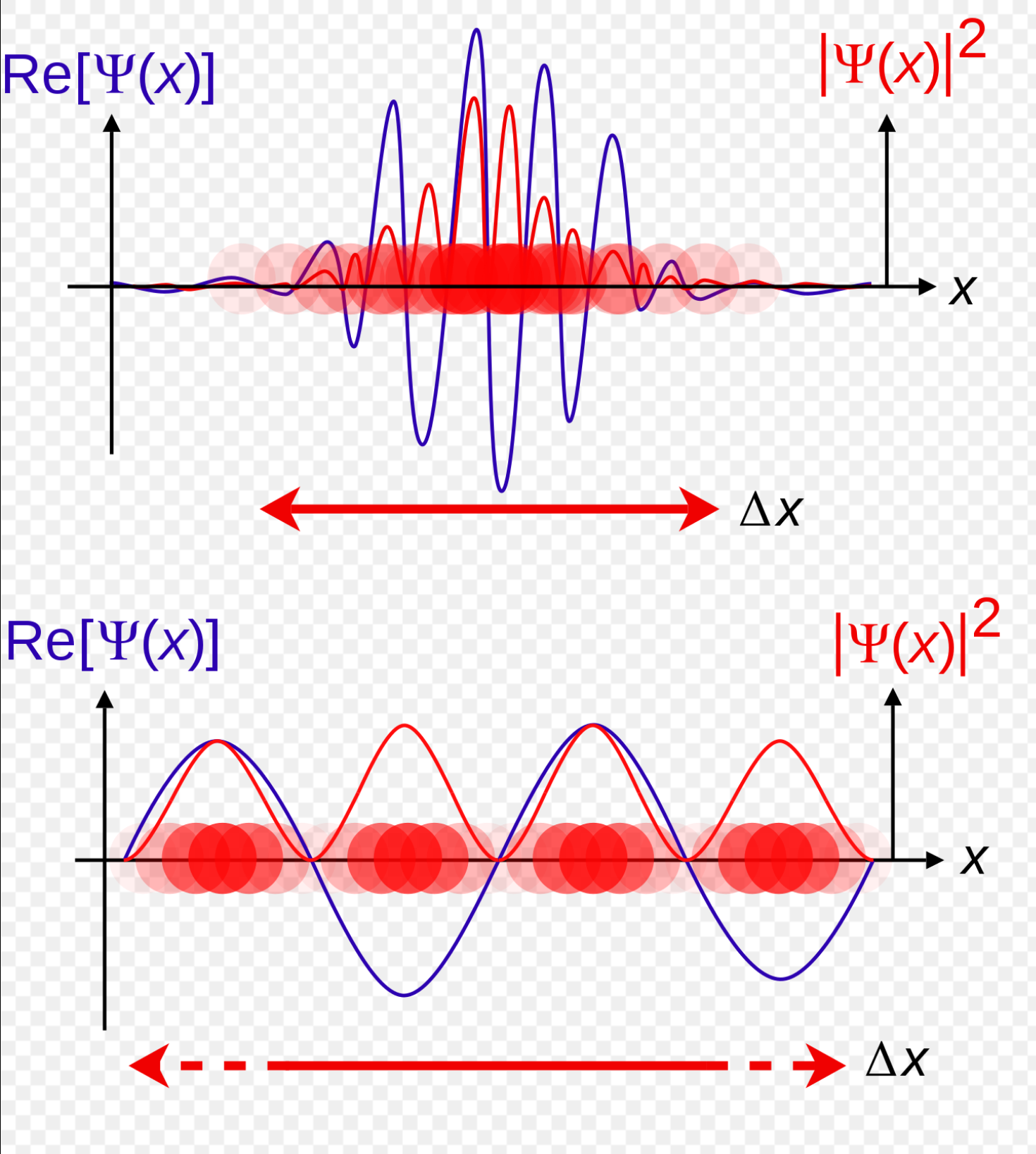 波函数实部(紫色),概率密度(红色)
波函数实部(紫色),概率密度(红色)
算子(Operator)
比如微分算子
量子力学算符
量子力学可观测量都可由算符表示,比如动能算符
基态(Ground state)
能量最低,体系最稳定的状态
固体中的多体问题
求解以下多体薛定谔方程,即可得到体系基态
\widehat{\boldsymbol{H}} \boldsymbol{\psi}\left(\left\{r_{i}\right\},\left\{R_{I}\right\}\right)=\boldsymbol{E} \boldsymbol{\psi}\left(\left\{r_{i}\right\},\left\{R_{I}\right\}\right)
其中,表示电子的位置,表示原子核的位置,为带电粒子库伦能以及动能算符的总和。 求解这样的问题是复杂的,为此给出以下近似。
Born Oppenheimer approximation(多体多电子)
原子核质量远大于电子质量。因此,电子运动比原子核快得多,可以将原子核运动和电子运动分离处理,先固定原子核,从而对波函数进行解耦,即
\psi\left(\left\{r_{i}\right\},\left\{R_{l}\right\}\right) \rightarrow \psi_{N}\left(\left\{R_{l}\right\}\right) * \psi_{e}\left(\left\{r_{i}\right\}\right)
于是,多体薛定谔方程可变为多电子薛定谔方程,即
\widehat{H} \psi\left(\boldsymbol{r}_{\mathbf{1}}, \boldsymbol{r}_{2}, \boldsymbol{r}_{3} \ldots \boldsymbol{r}_{N}\right)=E \psi\left(\boldsymbol{r}_{1}, \boldsymbol{r}_{2}, \boldsymbol{r}_{3} \ldots \boldsymbol{r}_{N}\right)
电子哈密顿量为
\widehat{\boldsymbol{H}}=-\frac{\hbar^{2}}{2 m_{e}} \sum_{i}^{N_{e}} \nabla_{i}^{2}+\sum_{i}^{N_{e}} V_{e x t}\left(r_{i}\right)+\sum_{i=1}^{N_{e}} \sum_{j>1} U\left(r_{i}, r_{j}\right)
其中,第一项表示电子动能,第二项表示电子和原子核相互作用能,第三项为电子和电子的相互排斥能。 即使暂时不考虑原子核,求解上述薛定谔方程仍是困难的,方程的维度是。
密度泛函理论 (波函数 电子密度)
电子密度定义
n(\boldsymbol{r})=\psi^{*}\left(\boldsymbol{r}_{1}, \boldsymbol{r}_{2} \ldots r_{N}\right) \psi\left(r_{1}, r_{2} \ldots r_{N}\right)
更进一步,对于多电子问题,可以认为某一电子是在其他电子产生的总电场中运动,那么多电子波函数可认为是
\psi\left(r_{1}, r_{2}, r_{3} \ldots r_{N}\right)=\psi\left(r_{1}\right) * \psi_{2}\left(r_{2}\right) * \psi_{3}\left(r_{3}\right) * \cdots * \psi_{N}\left(r_{N}\right)
对应的电子密度可写为
n(\boldsymbol{r})=2 \sum_i^{N_e} \psi_{i}^{*}(r) \psi_{i}(r)
从而多电子问题可以转化为单电子问题。
密度泛函理论基本定理
- 定理一 基态能量是电子密度的泛函,即。
- 最低能量对应的电子密度即为基态电子密度,即 为此,我们需要了解能量泛函的具体形式,如下
E\left[\left\{\psi_{i}\right\}\right]=E_{\text {known }}\left[\left\{\psi_{i}\right\}\right]+E_{X C}\left[\left\{\psi_{i}\right\}\right]
其中,已知表达式的泛函包括电子动能项、电子原子核相互作用项、电子电子相互作用项以及原子核原子核相互作用项,即
\begin{aligned}
&E_{\text {known }}\left[\left\{\psi_{i}\right\}\right]=-\frac{\hbar}{m_{e}} \sum_{i} \int \psi_{i}^{*} \nabla^{2} \psi_{i} d^{3} r+\int V(r) n(r) d^{3} r+\frac{e^{2}}{2} \iint \frac{n(r) n\left(r^{\prime}\right)}{r-r^{\prime}} d^{3} r d^{3} r^{\prime}+E_{i o n}
\end{aligned}
不存在解析表达式的能量泛函为交换关联泛函,该项包含所有的量子相互作用,常用的两种近似为LDA(Local density approximation) 和 GGA (Generalized gradient approximation)。
Kohn Sham Scheme
求解单电子波函数方程
\left[-\frac{\hbar}{2 m} \nabla^{2}+V(\boldsymbol{r})+V_{\mathrm{H}}(\mathbf{r})+\mathrm{V}_{\mathrm{XC}}(\mathbf{r})\right] \psi_{i}(\boldsymbol{r})=\epsilon_{i}(\boldsymbol{r}) \psi_{i}
哈密顿量中第一项为电子动能,第二项为电子原子核相互作用,第三项为电子与具有电子密度的电子气相互作用能,第四项为交换关联泛函。 基本算法流程为
- 给定初始电子密度;
- 求解单电子薛定谔方程得到波函数;
- 根据单电子波函数得到电子密度;
- 新的电子密度和上一步电子密度比较,判断是否收敛。
晶体和平面波
本文章使用limfx的vscode插件快速发布